


特别程序
根据《食品药品监管总局关于印发医疗器械生产企业分级分类监督管理规定的通知》(食药监械监〔2014〕234号)规定,申请材料齐全,符合法定形式,当场备案。备案后三个月内须开展一次全项目检查。
受理条件:
申请材料齐全,符合法定形式。
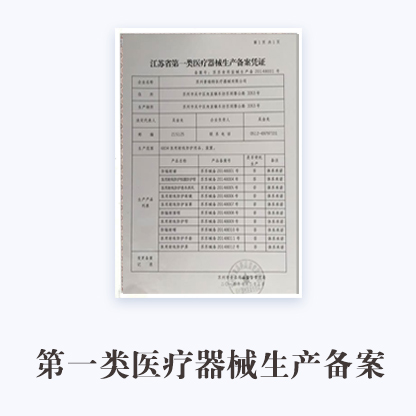
1第一类医疗器械生产备案表
2所生产产品的医疗器械备案凭证复印件
3经备案的产品技术要求复印件
4营业执照复印件
5法定代表人、企业负责人身份证明复印件
6生产、质量和技术负责人的身份、学历职称证明复印件
7生产场地的证明文件(有特殊生产环境要求的,还应提交设施、环境的证明文件)复印件
8质量手册和程序文件
9医疗器械生产质量管理规范自查表
10经办人授权证明
设定依据
【行政法规】《医疗器械监督管理条例》(国务院令第276号公布,国务院令第650号第一次修改,国务院令第680号第二次修改) 第二十一条 从事第一类医疗器械生产的,由生产企业向所在地设区的市级人民政府食品药品监督管理部门备案并提交其符合本条例第二十条规定条件的证明资料。 【规章】《医疗器械生产监督管理办法》(国家食品药品监督管理总局第7号令公布,国家食品药品监督管理总局第37号修改) 第十一条 开办第一类医疗器械生产企业的,应当向所在地设区的市级食品药品监督管理部门办理第一类医疗器械生产备案,提交备案企业持有的所生产医疗器械的备案凭证复印件和本办法第八条规定的资料(第二项除外)。 食品药品监督管理部门应当当场对企业提交资料的完整性进行核对,符合规定条件的予以备案,发给第一类医疗器械生产备案凭证。 第二十一条 第一类医疗器械生产备案凭证内容发生变化的,应当变更备案。 备案凭证遗失的,医疗器械生产企业应当及时向原备案部门办理补发手续。 第三十一条第二款 受托生产第一类医疗器械的,受托方应当依照本办法第二十一条的规定,向原备案部门办理第一类医疗器械生产备案变更。
扬州一类医疗器械生产代办区域:扬州市、广陵区、邗江区、江都区、仪征市、高邮市、宝应县




